关于本系列
本文为系列文章《中国生物医药企业拓展欧洲临床试验的监管挑战》的第一篇。该系列共八篇,将围绕欧洲临床试验监管体系的关键环节展开,帮助中国生物医药企业在拓展欧洲临床研究时,更顺利、更合规地应对各类法规要求。
引言
《欧盟临床试验法规》(EU Clinical Trials Regulation, EU CTR 536/2014)及其配套系统“临床试验信息系统”(Clinical Trials Information System, CTIS)正在重塑欧盟(EU)及欧洲经济区(EEA)范围内的临床试验审批与管理模式。对于尚无欧盟/欧洲经济区经验的生物技术或制药企业而言,了解并掌握这一监管框架,是推动临床试验顺利、合规开展的重要前提。
一、EU CTR 的核心作用
自2022年1月31日起全面实施的EU CTR,取代了以往的《临床试验指令》,建立了适用于所有欧盟及欧洲经济区成员国的统一流程。
其主要目标包括:
- 通过CTIS集中在线系统简化申报流程;
- 提升欧盟/欧洲经济区范围内临床试验的透明度与一致性;
- 加强受试者安全与监管。
这一统一化方法使申办方能够通过一次集中申报覆盖多个国家,从而减少重复质询及审批延迟的风险。
二、与中国 IND 体系的差异
与中国的药物临床试验申请(IND)流程不同,EU CTR 将集中科学评估(Part I)与各成员国伦理审查(Part II)相结合。
在中国,申办方通常向国家药品监督管理局(NMPA)提交 IND 申请,由国家药审中心(CDE)进行技术审评,伦理审查则由各试验机构分别开展,流程相对独立。
而在 EU CTR 框架下,Part I 的科学评估由欧盟成员国间协同完成,实现技术审评结果在多国间共享;Part II 的伦理审查则由各国伦理委员会依据本地要求独立进行。
这种“双层结构”要求申办方在文件一致性、语言翻译及时间规划方面提前布局与协调。
为更直观地理解两种体系在审评机制、时限与监管导向方面的差异,以下表格总结了中国 IND(结合 2025 年监管优化措施)与欧盟 EU CTR 的关键特点对比
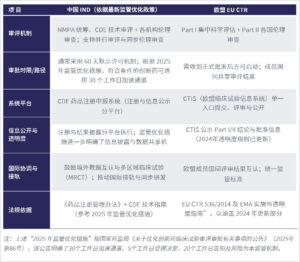
对于计划开展中欧多区域研究的申办方而言,了解这两种体系的审评逻辑与时间框架,是制定全球开发策略的重要前提。
三、成功申报的关键步骤
有效开展EU CTR申报的关键在于系统性规划与充分准备:
- 提前规划 Part I 与 Part II 的审评流程;
- 确保核心文件内容与 EU CTR、EMA 及 ICH GCP 要求保持一致——研究方案(Protocol)、研究者手册(Investigator’s Brochure, IB)、试验用药品资料(IMPD)应符合EU CTR、EMA及ICH GCP标准;
- 熟悉CTIS操作流程,包括权限分配、沟通机制与时间节点;
- 与具备中欧双向监管经验的CRO合作,以确保合规性与沟通效率。
充分的准备与专业支持对于首次使用CTIS的申办方尤为关键。
四、从合规到机遇
尽管适应EU CTR的过程可能较为复杂,但这一过程也有助于企业构建更强的全球合规能力与数据可信度。
深入理解并有效应用欧盟法规的企业,将能够加快审评进度、提升研究透明度,并在全球药物开发中建立长期竞争优势。
结语
EU CTR 为欧洲临床试验管理体系确立了新的统一标准。对于希望拓展国际化研发的生物医药创新企业而言,掌握EU CTR与CTIS流程,不仅关乎合规,更是通往高效、高质量全球研究的关键一步。
上一篇文章:中国生物医药企业拓展欧洲临床试验的监管挑战